Protein Kinases and Cancer Research
![]()
Protein kinase genes are signaling switches with a conserved catalytic domain that phosphorylate protein substrates and play a critical role in cell signaling pathways. A landmark for understanding the molecular basis of kinase function was the elucidation of the crystal structures of protein kinase A. Since this discovery, more than 1000 crystal structures of 119 unique human protein kinases have been solved, resulting in the growing wealth of structural knowledge about the kinase catalytic domain. The crystal structures have revealed considerable structural differences between closely related active and highly specific inactive kinase forms. Drug discovery against protein kinases has concentrated mainly on small molecules that target the ATP binding site of the conserved catalytic domain. However, with over 500 protein kinase genes identified in the human genome and the highly conserved ATP-binding site, a considerable effort is needed to design drugs that select for individual kinase members. A growing number of kinase inhibitors selectively target the inactive conformation whereas other compounds bind to both conformations with similar affinity. Inhibitors that bind to the inactive conformation face weaker competition from cellular ATP and may act primarily by shifting equilibrium between conformational states in a way that prevents kinase activation, rather than by inhibiting kinase activity directly. The complete sequencing of the human genome and high-throughput generationof genomic data have opened avenues for a systematic approachto understanding the complex biology of cancer and clinical targetingof activated oncogenes in cancer.
Our Research is focused on the development of integrated platforms of computational and experimental approaches for high-throughput ligand screening and design of kinase inhibitors.The intellectual merit of our research stems from a significant therapeutic position of the protein kinase family, which is ideally suited for this objective because of the growing wealth of structural and functional information about these genes. Many protein kinases have emerged as important therapeutic targets for combating diseases caused by abnormalities in signal transduction pathways. Protein kinases are the most common protein domains that are implicated in cancer and there are more than 500 encoded in the human genome. Rooted in statistical-mechanical description of biological systems and exploiting fundamental similarities between protein folding and molecular recognition, our approach seeks to establish a novel computational strategy for ligand screening and design of kinase inhibitors based on the energy landscape models of ligand-protein binding. In silico approaches are integrated with a carefully orchestrated funnel of experimental validation and design studies that include high throughput screening and biological analysis, chemical synthesis and library design, kinome-wide inhibitor profiling and biophysical characterization.
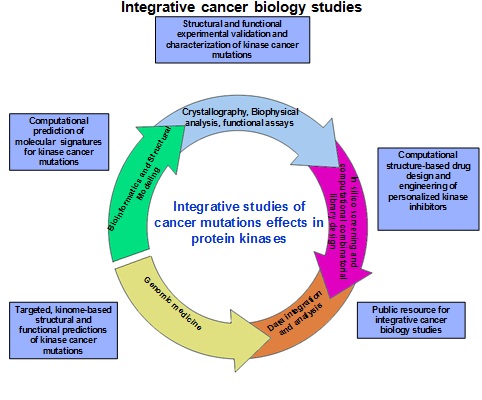
Our objective is to develop an integrated platform of validated computational approaches, models and tools to facilitate identification, prediction and functional characterization of molecular signatures of cancer-causing mutations that will enable the design of personalized cancer medicine to combat specific genomic profiles. The protein kinase family is an ideal family to achieve this objective because of the growing wealth of structural and functional information about these genes, as well as the prominent role that protein kinases play as therapeutic targets for cancer intervention.
Our goals are (a) integration of computational predictions and experimental validations of cancer mutations effects in therapeutically important protein kinase targets to provide a platform for structurally informed functional analysis of somatic mutations in the protein kinase genes; (b) structural and biophysical characterization of protein kinase dynamics, stability and binding in the normal and oncogenic states to enable targeted design of specific kinase inhibitors as well as reengineering and optimizing the clinical effects of existing drugs.
OUR PROJECTS
Development machine learning, structural bioinformatics and protein modeling approaches for prediction and characterization of molecular signatures of cancer mutations in protein kinases.
Integration of computational and experimental approaches to characterize structural and functional signatures of cancer mutations in the protein kinase genes.
Application the developed and validated computational models and approaches for structurally informed functional annotation and predictions of somatic mutation in the protein kinases genes.
In silico structure-based design of personalized kinase drugs to combat specific genomic profiles in kinase genes.
Development of a bioinformatics resource for integrative cancer biology studies and personalized drug design.
Tracing the effect of sequence variations responsible for phenotypic variations and providing insight into the molecular pathologic lesions associated with disease susceptibility can usher in an era of individualized medicine. These integrative cancer biology studies are undertaken to provide: (a) insights into molecular effects of cancer mutations in protein kinases underlying the biology of cancer; (b) practical benefits to cancer research. The results from experimental structural, biophysical and functional studies will be brought back to computational approaches for validation and refinement of the computational models. The insights provided by computational predictions will inform, guide and facilitate experimental studies of our collaborators exploring the molecular pathology of tumorigenesis and the design of personalized kinase cancer agents. To achieve the goals of this project we have assembled an integrated team of accomplished investigators and collaborators with complementary expertise and proven track records in the areas ranging from computational and structural biology to statistical genetics, functional genomics and chemical biology.